
Yolanda L. Li, Chih-Te Zee, Janice B. Lin, Victoria M. Basile, Mit Muni, Maria D. Flores, Julen Munárriz, Richard B. Kaner, Anastassia N. Alexandrova, K. N. Houk, Sarah H. Tolbert, and Yves Rubin “Fjord-Edge Graphene Nanoribbons with Site-Specific Nitrogen Substitution”, J. Am. Chem. Soc. 2020, 142, 18093–18102.
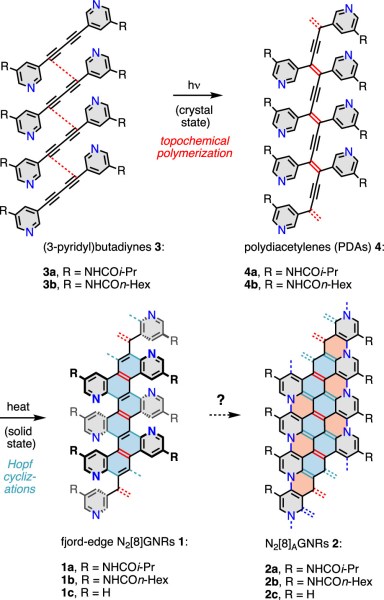
Abstract: The synthesis of graphene nanoribbons (GNRs) that contain site-specifically substituted backbone heteroatoms is one of the essential goals that must be achieved in order to control the electronic properties of these next generation organic materials. We have exploited our recently reported solid-state topochemical polymerization/cyclization-aromatization strategy to convert the simple 1,4-bis(3-pyridyl)butadiynes 3a,b into the fjord-edge nitrogen-doped graphene nanoribbon structures 1a,b (fjord-edge N2[8]GNRs). Structural assignments are confirmed by CP/MAS 13C NMR, Raman, and XPS spectroscopy. The fjord-edge N2[8]GNRs 1a,b are promising precursors for the novel backbone nitrogen-substituted N2[8]AGNRs 2a,b. Geometry and band calculations on N2[8]AGNR 2c indicate that this class of nanoribbons should have unusual bonding topology and metallicity.
Erik Peter Farr, Matthew T. Fontana, Chen-Chen Zho, Peiqi Wu, Yolanda L. Li, Nicholas Knutson, Yves Rubin, and Benjamin J. Schwartz “Bay-Linked Perylenediimides are Two Molecules in One: Insights From Ultrafast Spectroscopy, Temperature Dependence, and TD-DFT Calculations”, J. Phys. Chem. C. 2019, 123, 2127–2138.
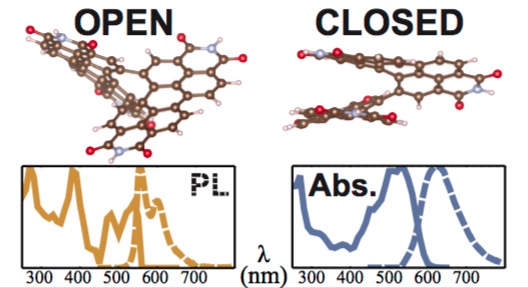
Abstract: Bay-linked di-perylenediimide (di-PDI) molecules are finding increasing use in organic electronics due to their steric hindrance that ‘twists’ the two monomer units relative to one another, decreasing molecular aggregation. In this paper we explore the electronic spectroscopy and ultrafast dynamics of the singly-linked β − β-S-di-PDI (2,9’- di(undecan-5-yl)-2’,9-di(undecan-6-yl)-[5,5’-bianthra[2,1,9-def:6,5,10-d’e’f’]diisoquinolin]- 1,1’,3,3’,8,8’,10,10’(2H,2’H,9H,9’H)-octaone). Excitation-emission spectroscopy reveals two distinct emitting species, which are further characterized by time-dependent density functional theory (TD-DFT), demonstrating that the bay-linked PDI dimers exist in two geometrical conformations. These conformations are an “open” geometry, where the two monomer sub-units are oriented nearly at right angles, giving them more J-like coupling, and a “closed” geometry, in which the two monomer sub-units are nearly π-stacked, resulting in more H-like coupling. Given the extent of through-space and through-bond coupling, however, neither di-PDI conformer can be well-described simply in terms of independently-coupled monomers; instead, a full quantum chemistry description is required to understand the electronic structure of this molecule. Temperature-dependent experiments and the TD-DFT calculations indicate that the “closed” conformer is ∼70 meV more stable than the “open” conformer, so that both conformers are important to the behavior of the molecule at room temperature and above. We use a combination of steady-state and femtosecond transient absorption and emission spectroscopies to globally fit the multiple electronic transitions underlying the spectra of both the “closed” and “open” conformers, which agree well with the TD-DFT calculations. The fact that di-PDI molecules are molecular species that adopt two distinct quasi-independent chemical identities has important ramifications for charge trapping and mobility in the organic electronic devices employing these materials.
Robert S. Jordan , Yolanda L. Li, Cheng-Wei Lin, Ryan D. McCurdy, Janice B. Lin , Jonathan L. Brosmer , Kristofer L. Marsh, Saeed I. Khan, K. N. Houk , Richard B. Kaner , and Yves Rubin* “Synthesis of N = 8 Armchair Graphene Nanoribbons from Four Distinct Polydiacetylenes”, J. Am. Chem. Soc. 2017, 139, 15878–15890.
UCLA Newsroom highlight: UCLA chemists synthesize narrow ribbons of graphene using only light and heat
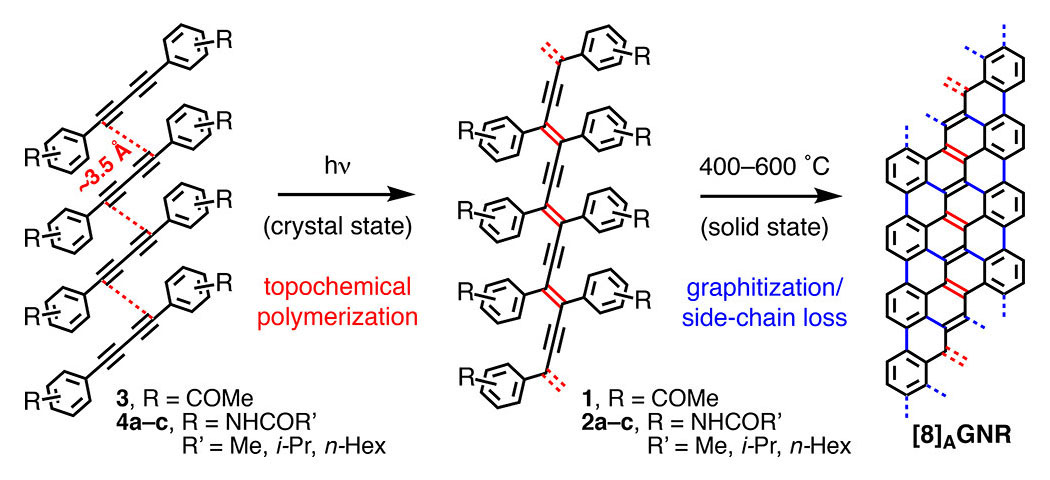
Abstract: We demonstrate a highly efficient thermal conversion of four differently substituted polydiacetylenes (PDAs 1 and 2a–c) into virtually indistinguishable N = 8 armchair graphene nanoribbons ([8]AGNR). PDAs 1 and 2a–c are themselves easily accessed through photochemically initiated topochemical polymerization of diynes 3 and 4a–c in the crystal. The clean, quantitative transformation of PDAs 1 and 2a–c into [8]AGNR occurs via a series of Hopf pericyclic reactions, followed by aromatization reactions of the annulated polycyclic aromatic intermediates, as well as homolytic bond fragmentation of the edge functional groups upon heating up to 600 °C under an inert atmosphere. We characterize the different steps of both processes using complementary spectroscopic techniques (CP/MAS 13C NMR, Raman, FT-IR, and XPS) and high-resolution transmission electron microscopy (HRTEM). This novel approach to GNRs exploits the power of crystal engineering and solid-state reactions by targeting very large organic structures through programmed chemical transformations. It also affords the first reported [8]AGNR, which can now be synthesized on a large scale via two operationally simple and discrete solid-state processes..
Robert S. Jordan and Yves Rubin* “The Allure of Metallic Stripes: Single-Sized Narrow Ribbons of Graphene”, Chem 2017, 2, 11–19.
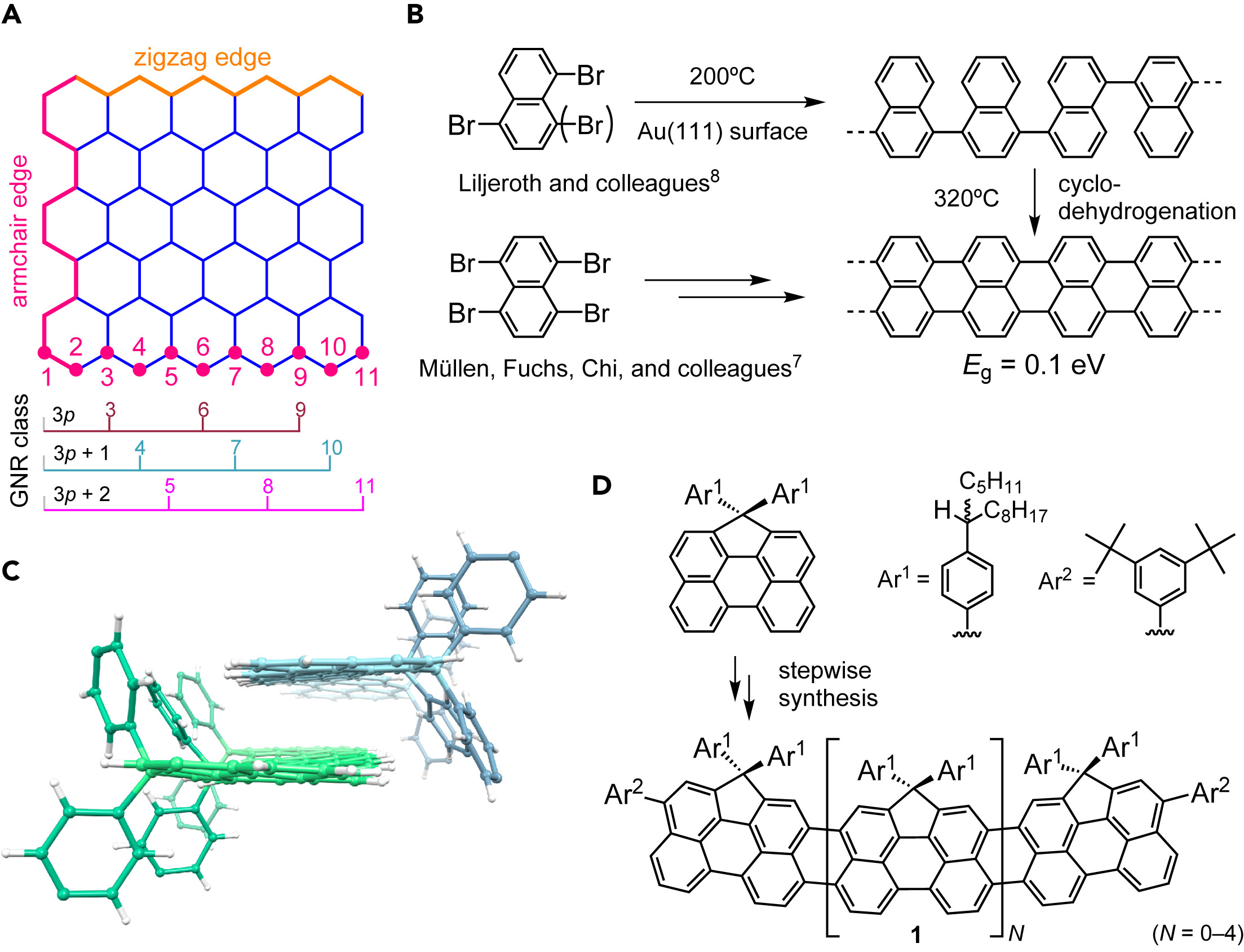
Preview article for: Wangdong Zeng, Hoa Phan, Tun Seng Herng, Tullimilli Y. Gopalakrishna, Naoki Aratani, Zebing Zeng, Hiroko Yamada, Jun Ding, Jishan Wu* “Rylene Ribbons with Unusual Diradical Character”, Chem 2017, 2, 81–92.
Abstract: Cutting-edge synthesis is gradually delivering new types of graphene nanoribbons, enabling comparison of their properties to theoretical predictions. In this issue of Chem, Wu and coworkers describe oligomers of N = 5 armchair graphene nanoribbons possessing near-metallic (Eg = 0.21 eV) behavior.
Robert S. Jordan, Yue Wang, Ryan D. McCurdy, Michael T. Yeung, Kristofer L. Marsh, Saeed I. Khan, Richard B. Kaner, and Yves Rubin* “Synthesis of Graphene Nanoribbons via the Topochemical Polymerization and Subsequent Aromatization of a Diacetylene Precursor”, Chem 2016, 1, 78–90.
Preview article: Paul J. Evans, Nazario Martín* “Graphene Nanoribbons via Crystal Engineering”, Chem 2016, 1, 16–18.
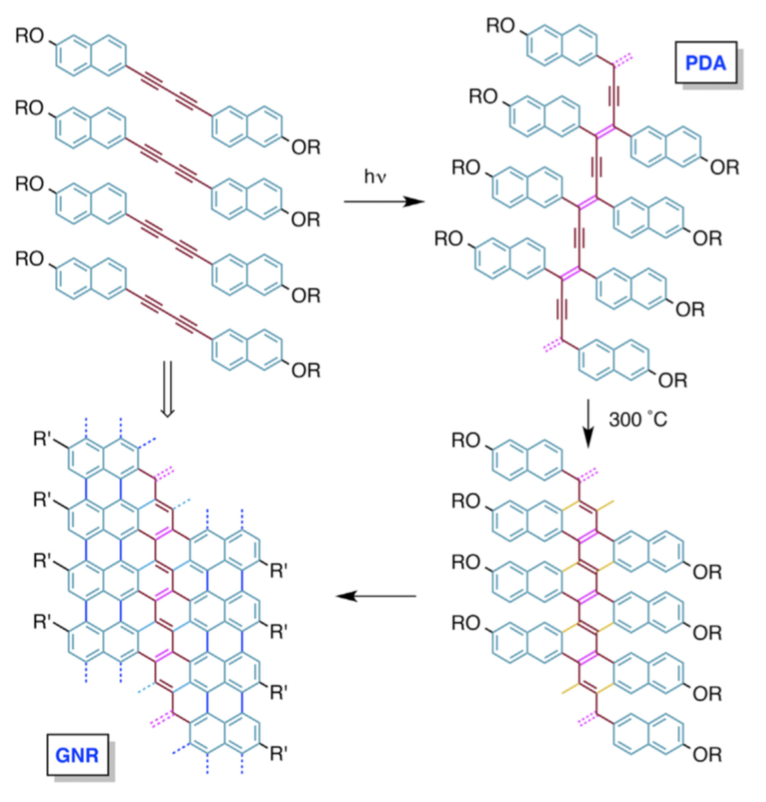
Abstract: We have developed a synthetic approach to graphene nanoribbons by using bu- tadiyne-containing monomers that are initially converted to polydiacetylenes via topochemical polymerization in the crystal. Subsequent aromatization of the isolated polydiacetylenes at surprisingly mild temperatures affords gra- phene nanoribbons $1.36 nm in width with a 1.4 eV bandgap. These transforma- tions take place solely in the solid state, in contrast to published on-surface or in-solution methods. This synthetic approach is well suited for electronic-device fabrication processes because it requires only UV light or heating and no external chemical reagents.
Mitsuharu Suzuki, Juliet F. Khosrowabadi Kotyk, Saeed I. Khan, and Yves Rubin* “Directing the Crystallization of Dehydro[24]annulenes into Supramolecular Nanotubular Scaffolds”, J. Am. Chem. Soc. 2016, 138, 5939–5956.

Abstract: The self-assembly of a series of dehydro[24]annulene derivatives into columnar stacks has been examined for its latent ability to form π-conjugated carbon-rich nanotubular structures through topochemical polymerizations. We have studied the parameters affecting self-assembly, including the nature of the substituent and crystallization conditions, using 10 different dehydro[24]annulene derivatives. In particular, hydrogen-bonding interactions through carbamate groups were found to be especially useful at directing the formation of nanotubular supramolecular assemblies. We have also evaluated the electronic coupling between neighboring dehydroannulene molecules within these supramolecular assemblies. Density functional calculations on the stacked supramolecular nanotube assemblies show that transfer integrals vary considerably between the three columnar assemblies, ranging from moderate to high (59–98 meV for the highest occupied molecular orbitals, 63–97 meV for the lowest unoccupied molecular orbitals), depending on the local molecular topology. In addition, the dehydro[24]annulene derivatives afforded distinct architectures in the crystal, including nanochannel arrays, sheets with solvent-filled pores, and lamellae. This work is an essential step toward a controlled formation of covalently linked carbon-rich nanostructures generated from molecular precursors with a latent diacetylene reactivity.
Suzuki, M.; Guo, Z.; Tahara, K.; Khosrowabadi Kotyk, J. F.; Nguyen, H.; Gotoda, J.; Iritani, K.; Rubin, Y.; Tobe, Y.* “Self-Assembled Dehydro[24]annulene Monolayers at the Liquid/Solid Interface: Towards On-Surface Synthesis of Tubular π-Conjugated Nanowires”, Langmuir 2016, 32, 5532–5541.
![self-assembled [24]annulenes](https://rubingroup.org/wp-content/uploads/2011/02/self-assembled-24annulenes.jpg)
Abstract: We have studied the self-assembly behavior of dehydro[24]annulene (D24A) derivatives 1, 2a–d, and 3a–c at the liquid/solid interface using scanning tunneling microscopy (STM). Both the relative placement and the nature of the four D24A substituents strongly influence the self-assembly pattern. Overall, the eight D24A derivatives examined in this study display seven types of 2D packing patterns. The D24A derivatives 1, 2a, and 3a have either two or four stearate groups, and adopt face-on configurations of their macrocyclic cores with respect to the highly oriented pyrolytic graphite (HOPG) surface. Their 2D packing pattern is determined by the interchain spacings and number of stearate substituents. The D24A derivatives 2b–2d and 3b–3c bear hydrogen-bonding carbamate groups to further strengthen intermolecular interactions. Face-on patterns were also observed for most of these compounds, while an unstable edge-on self-assembly was observed in the case of 2b at room temperature. Stable edge-on self-assemblies of D24A derivatives were sought for this work as an important stepping-stone to achieving the on-surface topochemical polymerization of these carbon-rich macrocycles into tubular π-conjugated nanowires. The overall factors determining the 2D packing patterns of D24As at the liquid/solid interface are discussed on the basis of theoretical simulations, providing useful guidelines for controlling the self-assembly pattern of future D24A macrocycles.
Huber, R. C.; Ferreira, A. S.; Aguirre, J. C.; Kilbride, D.; Toso, D. B.; Mayoral, K.; Zhou, Z. H.; Kopidakis, N.; Rubin, Y.; Schwartz, B. J.; Mason, T. G.; Tolbert, S. H.* “Structure and Conductivity of Semiconducting Polymer Hydrogels”, J. Phys. Chem. B 2016, 120, 6215–6224.
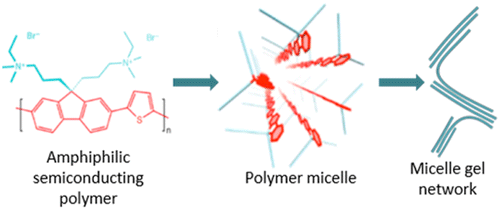
Abstract: Poly(fluorene-alt-thiophene) (PFT) is a conjugated polyelectrolyte that self-assembles into rod-like micelles in water, with the conjugated polymer backbone running along the length of the micelle. At modest concentrations (∼10 mg/mL in aqueous solutions), PFT forms hydrogels, and this work focuses on understanding the structure and intermolecular interactions in those gel networks. The network structure can be directly visualized using cryo electron microscopy. Oscillatory rheology studies further tell us about connectivity within the gel network, and the data are consistent with a picture where polymer chains bridge between micelles to hold the network together. Addition of tetrahydrofuran (THF) to the gels breaks those connections, but once the THF is removed, the gel becomes stronger than it was before, presumably due to the creation of a more interconnected nanoscale architecture. Small polymer oligomers can also passivate the bridging polymer chains, breaking connections between micelles and dramatically weakening the hydrogel network. Fits to solution-phase small-angle X-ray scattering data using a Dammin bead model support the hypothesis of a bridging connection between PFT micelles, even in dilute aqueous solutions. Finally, time-resolved microwave conductivity measurements on dried samples show an increase in carrier mobility after THF annealing of the PFT gel, likely due to increased connectivity within the polymer network.
Shaohua Huang, Guangye Zhang, Nicholas S. Knutson, Matthew T. Fontana, Rachel C. Huber, Amy S. Ferreira, Sarah H. Tolbert, Benjamin J. Schwartz, and Yves Rubin* “Beyond PCBM: Methoxylated 1,4-Bisbenzyl[60]fullerene Adducts for Efficient Organic Solar Cells”, J. Mater. Chem. A 2016, 4, 416-424.
Royal Society of Chemistry HOT Article: 2015 Journal of Materials Chemistry A Hot Papers
Abstract: Organic solar cells have been based mostly on conjugated polymers and the classic fullerene derivative PCBM and are characterized by modest open circuit voltages (Voc). Increasing Voc requires fullerene acceptors with higher LUMOs than PCBM. To date, most fullerene derivatives synthesized for this purpose either do not achieve the high photocurrent afforded by PCBM or show relatively poor compatibility with the next-generation low bandgap conjugated polymers used in high-efficiency organic solar cells. Here, we report the facile synthesis of methoxylated 1,4-bisbenzyl fullerene adducts and their application as efficient electron acceptors in conjugated polymer-based solar cells. The methoxy groups are found to be essential to increasing the LUMO levels, and accordingly the Voc, of the devices compared to the parent 1,4-bisbenzyl fullerene, and more importantly, to PCBM. The best fullerene 1,4-bisadduct provides a ∼20% enhancement in power conversion efficiency over PCBM when used with the classic crystalline polymer P3HT. When used in combination with a higher-performance low bandgap polymer, PTB7, the bisadduct both increases the device open-circuit voltage and maintains the high photocurrent provided by the more traditional PCBM. We also examine 10 different 1,4-fullerene bisadducts and show that the photovoltaic device performance is strongly influenced by the number and relative position of the methoxy substituents on the benzyl addends: moving a single methoxy substituent by one position on the benzyl rings can change the device efficiency by over a factor of 2.
Yue Wang, Jaime A. Torres, Adam Z. Stieg, Shan Jiang, Michael T. Yeung, Yves Rubin, Santanu Chaudhuri, Xiangfeng Duan, and Richard B. Kaner* “Graphene-Assisted Solution Growth of Vertically Oriented Organic Semiconducting Single Crystals”, ACS Nano, 2015, 9, 9486–9496.
UCLA Newsroom highlight: Scientists grow organic semiconductor crystals vertically for first time
Abstract: Vertically oriented structures of single crystalline conductors and semiconductors are of great technological importance due to their directional charge carrier transport, high device density, and interesting optical properties. However, creating such architectures for organic electronic materials remains challenging. Here, we report a facile, controllable route for producing oriented vertical arrays of single crystalline conjugated molecules using graphene as the guiding substrate. The arrays exhibit uniform morphological and crystallographic orientations. Using an oligoaniline as an example, we demonstrate this method to be highly versatile in controlling the nucleation densities, crystal sizes, and orientations. Charge carriers are shown to travel most efficiently along the vertical interfacial stacking direction with a conductivity of 12.3 S/cm in individual crystals, the highest reported to date for an aniline oligomer. These crystal arrays can be readily patterned and their current harnessed collectively over large areas, illustrating the promise for both micro- and macroscopic device applications.
Huber, R. C.; Ferreira, A. S.; Thompson, R.; Kilbride, D.; Knutson, N. S.; Sudha Devi, L.; Toso, D. B.; Zhou, Z. H.; Rubin, Y.*; Schwartz, B. J.*; Tolbert, S. H.* “Long-Lived Photoinduced Polaron Formation in Conjugated Polyelectrolyte/Fullerene Assemblies”, Science 2015, 348, 1340-1343.
UCLA Newsroom highlight: UCLA chemists devise technology that could transform solar energy storage
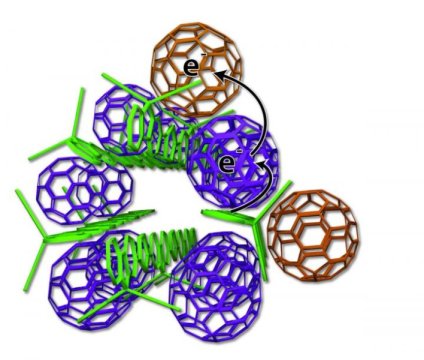
Abstract: The efficiency of biological photosynthesis results from the exquisite organization of photoactive elements that promote rapid movement of charge carriers out of a critical recombination range. If synthetic organic photovoltaic materials could mimic this assembly, charge separation and collection could be markedly enhanced. We show that micelle-forming cationic semiconducting polymers can coassemble in water with cationic fullerene derivatives to create photoinduced electron-transfer cascades that lead to exceptionally long-lived polarons. The stability of the polarons depends on the organization of the polymer-fullerene assembly. Properly designed assemblies can produce separated polaronic charges that are stable for days or weeks in aqueous solution.
The paper above was reviewed by UCLA’s Science Daily and by a number of scientific news sites.
Yamada, M.; Muto, Y.; Kurihara, H.; Slanina, Z.; Suzuki, M.; Maeda, Y.; Rubin, Y.; Olmstead, M.; Balch, A.; Nagase, S.; Lu, X.; Akasaka, T.* “Regioselective Cage Opening of La2@D2(10611)-C72 with 5,6-Diphenyl-3-(2-pyridyl)-1,2,4-triazine”, Angew. Chem. Int. Ed. 2014, 54, 2232–2235.
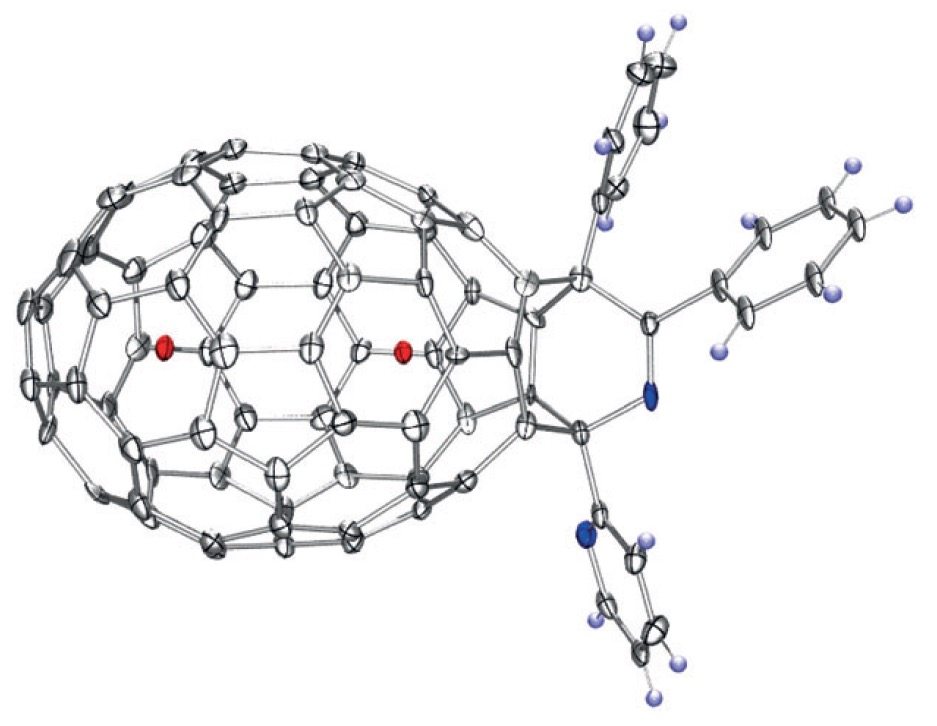
Abstract: The thermal reaction of the endohedral metallofullerene La2@D2(10611)-C72, which contains two pentalene units at opposite ends of the cage, with 5,6-diphenyl-3-(2-pyridyl)-1,2,4-triazine, proceeded selectively to afford only two bisfulleroid isomers. The molecular structure of one isomer was determined using single-crystal X-ray crystallography. The results suggest that the [4+2] cycloaddition was initiated in a highly regioselective manner at the C–C bond connecting two pentagon rings of C72. Subsequent intramolecular electrocyclization followed by cycloreversion resulted in the formation of an open-cage derivative having three seven-membered ring orifices on the cage and a significantly elongated cage geometry. The reduction potentials of the open-cage derivatives were similar to those of La2@D2-C72 whereas the oxidation potentials were shifted more negative than those of La2@D2-C72. These results point out that further oxidation could occur easily in the derivatives.
Aguirre, J. C.; Ferreira, A.; Ding, H.; Jenekhe, S. A.; Kopidakis, N.; Asta, M.; Pilon, L.; Rubin, Y.; Tolbert, S. H.; Schwartz, B. J.; Dunn, B.; Ozolins, V. “Panoramic View of Electrochemical Pseudocapacitor and Organic Solar Cell Research in Molecularly Engineered Energy Materials (MEEM)”, J. Phys. Chem. C 2014, 118, 19505–19523.
Abstract: Our program on capacitive energy storage is a comprehensive one that combines experimental and computational components to achieve a fundamental understanding of charge storage processes in redox-based materials, specifically transition metal oxides. Some of the highlights of this program are the identification of intercalation pseudocapacitance in Nb2O5, which enables high energy density to be achieved at high rates, and the development of a new route for synthesizing mesoporous films in which preformed nanocrystal building blocks are used in combination with polymer templating. The resulting material architectures have large surface areas and enable electrolyte access to the redox active pore walls, while the interconnected mesoporous film provides good electronic conductivity. Select first-principles density-functional theory studies of prototypical pseudocapacitor materials are reviewed, providing insight into the key physical and chemical features involved in charge transfer and ion diffusion. Rigorous multiscale physical models and numerical tools have been developed and used to reproduce electrochemical properties of carbon-based electrochemical capacitors with the ultimate objective of facilitating the optimization of electrode design. For the organic photovoltaic (OPV) program, our focus has been ongoing beyond the trial-and-error Edisonian approaches that have been responsible for the increase in power conversion efficiency of blend-cast (BC) bulk heterojunction blends of polymers and fullerenes. Our first approach has been to use molecular self-assembly to create the ideal nanometer-scale architecture using thermodynamics rather than relying on the kinetics of spontaneous phase segregation. We have created fullerenes that self-assemble into one-dimensional stacks and have shown that use of these self-assembled fullerenes lead to dramatically enhanced OPV performance relative to fullerenes that do not assemble. We also have created self-assembling conjugated polymers that form gels based on electrically continuous cross-linked micelles in solution, opening the possibility for water-processable “green” production of OPVs based on these materials. Our second approach has been to avoid kinetic control over phase separation by using a sequential processing (SqP) technique to deposit the polymer and fullerene materials in separate deposition steps. The polymer layer is deposited first, using solvents and deposition conditions that optimize the polymer crystallinity for swelling and hole mobility. The fullerene layer is then deposited in a second step from a solvent that swells the polymer but does not dissolve it, allowing the fullerene to penetrate into the polymer underlayer to the desired degree. Careful comparison of composition- and thickness-matched BC and SqP devices shows that SqP not only produces more efficient devices but also leads to devices that behave more consistently.
Aguirre, J. C.; Arntsen, C.; Hernandez, S.; Huber, R.; Nardes, A. M.; Halim, M.; Kilbride, D.; Rubin, Y.; Tolbert, S. H.; Kopidakis, N.; Schwartz, B. J.*; Neuhauser, D. “Understanding Local and Macroscopic Electron Mobilities in the Fullerene Network of Conjugated Polymer-based Solar Cells: Time-Resolved Microwave Conductivity and Theory”, Adv. Funct. Mat. 2014, 24, 784–792.
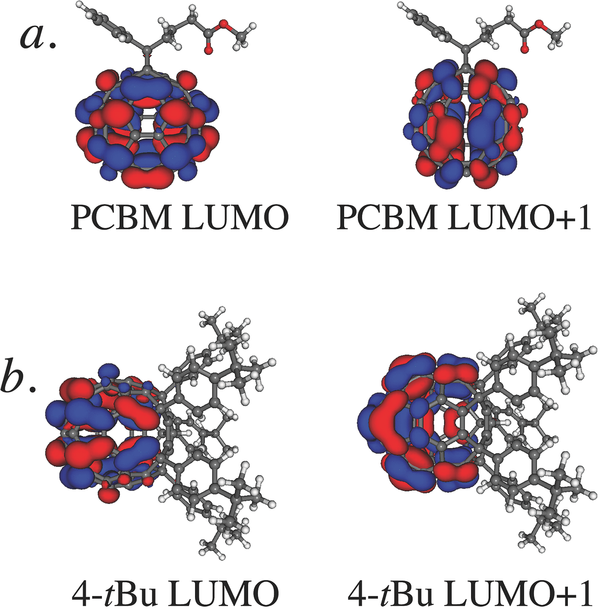
Abstract: The efficiency of bulk heterojunction (BHJ) organic photovoltaics is sensitive to the morphology of the fullerene network that transports electrons through the device. This sensitivity makes it difficult to distinguish the contrasting roles of local electron mobility (how easily electrons can transfer between neighboring fullerene molecules) and macroscopic electron mobility (how well-connected is the fullerene network on device length scales) in solar cell performance. In this work, a combination of density functional theory (DFT) calculations, flash-photolysis time-resolved microwave conductivity (TRMC) experiments, and space-charge-limit current (SCLC) mobility estimates are used to examine the roles of local and macroscopic electron mobility in conjugated polymer/fullerene BHJ photovoltaics. The local mobility of different pentaaryl fullerene derivatives (so-called ‘shuttlecock’ molecules) is similar, so that differences in solar cell efficiency and SCLC mobilities result directly from the different propensities of these molecules to self-assemble on macroscopic length scales. These experiments and calculations also demonstrate that the local mobility of phenyl-C60 butyl methyl ester (PCBM) is an order of magnitude higher than that of other fullerene derivatives, explaining why PCBM has been the acceptor of choice for conjugated polymer BHJ devices even though it does not form an optimal macroscopic network. The DFT calculations indicate that PCBM’s superior local mobility comes from the near-spherical nature of its molecular orbitals, which allow strong electronic coupling between adjacent molecules. In combination, DFT and TRMC techniques provide a tool for screening new fullerene derivatives for good local mobility when designing new molecules that can improve on the macroscopic electron mobility offered by PCBM.
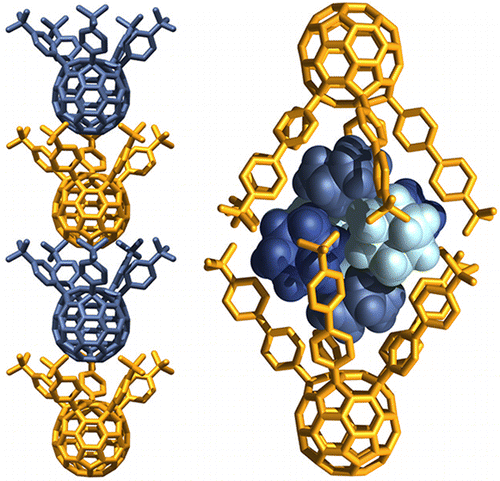
Clark, A. P.-Z.; Shi, C.; Ng, B. C.; Wilking, J. N.; Ayzner, A. L.; Stieg, A. Z.; Schwartz, B. J.; Mason, T. G.; Rubin, Y.*; Tolbert, S. H.* “Self-Assembling Semiconducting Polymers—Rods and Gels from Electronic Materials”, ACS Nano 2013, 7, 962–977.

Abstract: In an effort to favor the formation of straight polymer chains without crystalline grain boundaries, we have synthesized an amphiphilic conjugated polyelectrolyte, poly(fluorene-alt-thiophene) (PFT), which self-assembles in aqueous solutions to form cylindrical micelles. In contrast to many diblock copolymer assemblies, the semiconducting backbone runs parallel, not perpendicular, to the long axis of the cylindrical micelle. Solution-phase micelle formation is observed by X-ray and visible light scattering. The micelles can be cast as thin films, and the cylindrical morphology is preserved in the solid state. The effects of self-assembly are also observed through spectral shifts in optical absorption and photoluminescence. Solutions of higher-molecular-weight PFT micelles form gel networks at sufficiently high aqueous concentrations. Rheological characterization of the PFT gels reveals solid-like behavior and strain hardening below the yield point, properties similar to those found in entangled gels formed from surfactant-based micelles. Finally, electrical measurements on diode test structures indicate that, despite a complete lack of crystallinity in these self-assembled polymers, they effectively conduct electricity.
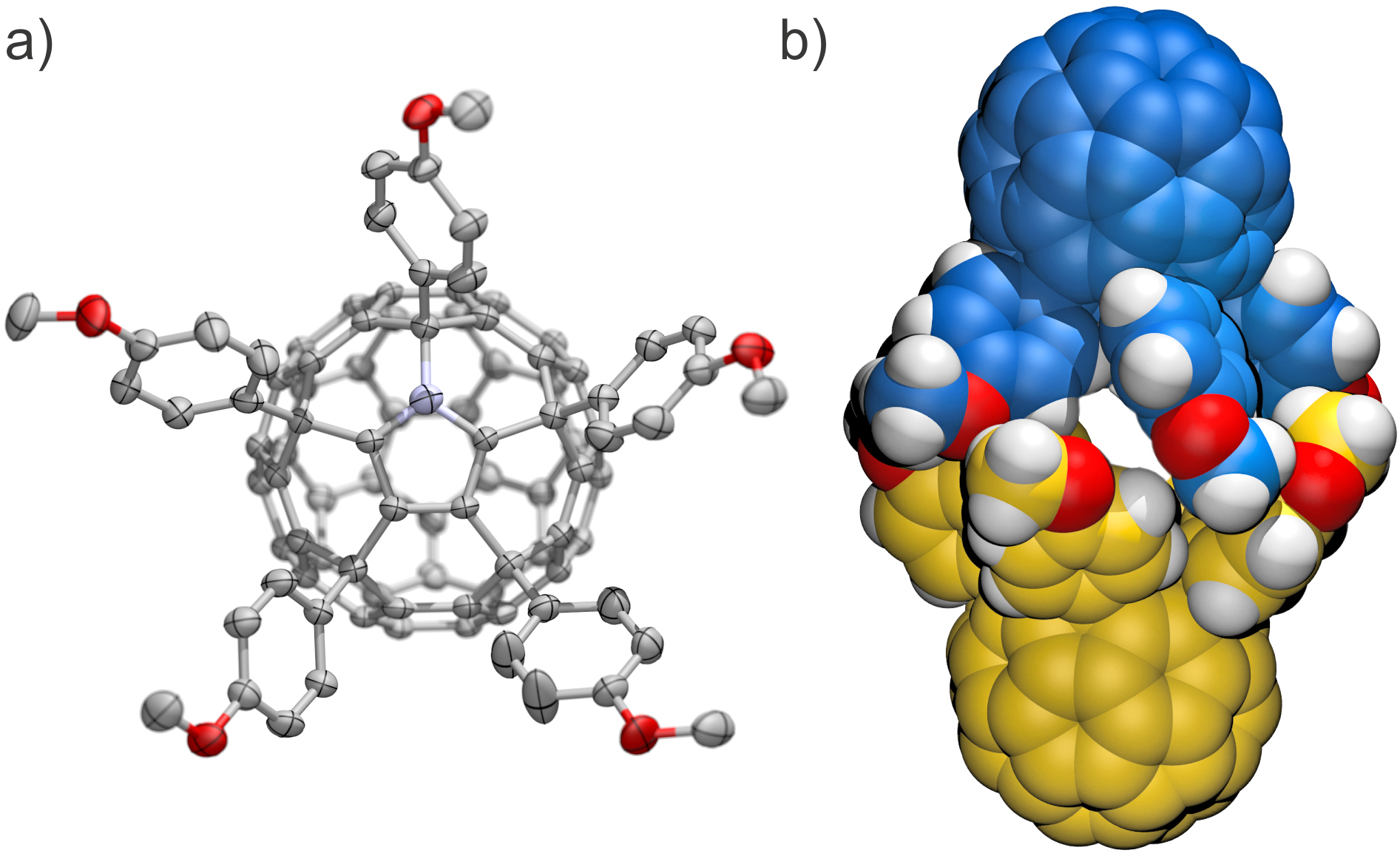
Neubauer, R.; Hampel, F.; Rubin, Y.; Hirsch, A.* “Pentaarylazafullerenes and their Triaryldihydro and Tetraarylmonohydro Precursors”, Chem. Eur. J. 2012, 51, 11722 –11726.
Abstract: A family of pentaaryl[C59N]azafullerenes and their mono- and dihydro precursors was isolated by acid catalyzed reaction of a C59N precursor with electron-rich aromatic compounds. Single-crystal X-ray diffraction on two compounds reveals characteristic packing motifs; the triaryl dihydro derivative has a pseudo-stacked, feather-in-cavity arrangement shown at left.
![Crystal-Packing Trends for a Series of 6,9,12,15,18-Pentaaryl-1-hydro[60]fullerenes](https://rubingroup.org/wp-content/uploads/2011/02/shuttlecock_x-ray_structures.gif)
Kennedy, R. D.; Halim, M.; Khan, S.I.; Schwartz, B.; Tolbert, S.; Rubin, Y.* “Crystal-Packing Motifs for a Series of 6,9,12,15,18-Pentaaryl-1-hydro[60]fullerenes”, Chem. Eur. J. 2012, 18, 7418–7433.
Abstract: The relationship between the size of the substituents of aryl groups in a series of fifteen 6,9,12,15,18-pentaaryl-1-hydro[60]fullerenes and the solid state structures and packing motifs of these compounds has been analyzed. Pentaarylfullerenes have a characteristic ‘badminton shuttlecock’ shape which causes several derivatives to crystallize into columnar stacks. However, many pentaarylfullerenes form non-stacked structures with, for example, dimeric, layered, diamondoid, or feather-in-cavity relationships between molecules. Computational modeling gave a qualitative estimate of the best shape match between the ball and socket surfaces of each pentaarylfullerene. The best match was for pentaarylfullerenes having large, spherically shaped para-substituents on the aryl groups. The series of pentaarylfullerenes was characterized by single-crystal X-ray diffraction. A total of 34 crystal structures were obtained as various solvates and were categorized by their packing motifs.
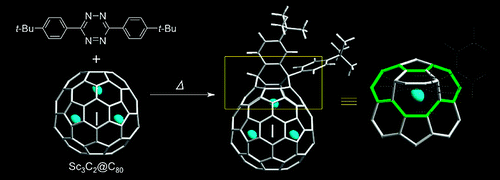
Kurihara, H.; Iiduka, Y.; Rubin, Y.; Waelchli, M.; Mizorogi, N.; Slanina, Z.; Tsuchiya, T.; Nagase, S.; Akasaka, T.* “Unexpected Formation of a Sc3C2@C80 Bisfulleroid Derivative”, J. Am. Chem. Soc. 2012, 134, 4092–4095.
Abstract: The reaction of tetrazine 1 with Sc3C2@C80 exclusively affords the open-cage derivative 2 instead of the expected C2-inserted derivative 3 bearing a four-membered ring, as previously obtained for C60. The structure of 2 has been firmly established by NMR spectroscopy and theoretical calculations. EPR spectroscopy shows that a single Sc atom of the Sc3C2 cluster gets located within the bulge created by the bridging addend, which is a first step toward release of the internal metal atoms.
Tassone, C.; Ayzner, A.; Kennedy, R. D.; Halim, M.; So, M.; Rubin, Y.*; Tolbert, S.*; Schwartz, B.* “Using Pentaarylfullerenes to Understand Network Formation in Conjugated Polymer-Based Bulk-Heterojunction Solar Cells”, J. Phys. Chem. C, 2011, 115, 22563–22571.
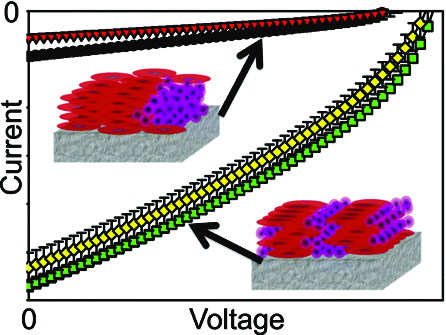
Abstract: We have synthesized a series of pentaarylfullerene derivatives and incorporated them into bulk-heterojunction (BHJ) solar cells using poly(3-hexylthiophene) as the electron donating material. Through a combination of grazing incidence X-ray diffraction and atomic force microscopy we see two distinct nanoscale morphologies emerge across the series. Investigating the device physics using J–V characterization, light intensity dependence, and fluorescence quenching studies, we are able to correlate the morphology to the device physics. This has afforded the opportunity to clearly see how altering the nanoscale phase segregation can change the distinct physical processes occurring within a BHJ solar cell. We observe an order of magnitude increase in the efficiency of devices that utilize self-assembling fullerene derivatives over their nonassembled counterparts. More importantly, however, these results allow us to shed light on the mechanism behind this differential phase segregation using a fullerene self-assembly model. On the basis of the data, we propose new design rules for the structure of future generations of fullerene electron accepting materials for use in BHJ solar cells.
Halim, M.; Kennedy, R. D.; Suzuki, M.; Khan, S. I.; Diaconescu, P. L.; Rubin, Y.* “Complexes of Gold(I), Silver(I), and Copper(I) with Pentaaryl[60]fullerides”, J. Am. Chem. Soc. 2011, 133, 6841–6851.

Abstract: Gold(I), silver(I), and copper(I) phosphine complexes of 6,9,12,15,18-pentaaryl[60]fullerides 1a and 1b, namely, [(4-MeC6H4)5C60]Au(PPh3) (2a), [(4-t-BuC6H4)5C60]Au(PPh3) (2b), [(4-MeC6H4)5C60]Ag(PCy3) (3a), [(4-t-BuC6H4)5C60]Ag(PPh3) (3b), [(4-t-BuC6H4)5C60]Ag(PCy3) (3c), [(4-MeC6H4)5C60]Cu(PPh3) (4a), and [(4-t-BuC6H4)5C60]Cu(PPh3) (4b), have been synthesized and characterized spectroscopically. All complexes except for 3c were also characterized by single-crystal X-ray diffraction. Several coordination modes between the cyclopentadienyl ring embedded in the fullerene and the metal centers are observed, ranging from η1 with a slight distortion toward η3 in the case of gold(I), to η2/η3for silver(I), and η5 for copper(I). Silver complexes 3a and 3b are rare examples of crystallographically characterized Ag(I) cyclopentadienyls whose preparation was possible thanks to the steric shielding provided by fullerides 1a and 1b, which stabilizes these complexes. Silver complexes3a and 3b both display unexpected coordination of the cyclopentadienyl portion of the fulleride anion with Ag(I). DFT calculations on the model systems (H5C60)M(PH3) and CpMPH3 (M = Au, Ag, or Cu) were carried out to probe the geometries and electronic structures of these metal complexes.
Suzuki, M.; Comito, A.; Khan, S. I.; Rubin, Y.* “Planarization of a Dehydro-[24]annulene by Noncovalent Interactions in the Crystal”, Org. Lett. 2010, 12, 2346–2349.

Abstract: Dehydro[24]annulene 1c adopts an unusual planarized conformation in the crystal. A multilayered stack of hydrogen-bonded grids delineates tightly packed nanotubular channels. The related macrocycles 1a and 1b, on the other hand, have the expected puckered conformations in the crystal.
Halim, M.; Kennedy, R. D.; Khan, S.I.; Rubin, Y.* “Gold(I) Triphenylphosphine Complexes Incorporating Pentaarylfulleride Ligands”, Inorg. Chem. 2010, 49, 3974–3976.

Abstract: Two gold(I) complexes, (Ph3P)Au[C60(4-MeC6H4)5] (1) and (Ph3P)Au[C60(4-t-BuC6H4)5] (2), were prepared in excellent yield and characterized by single-crystal X-ray diffraction. Complex 1, grown from two solvent systems, has different coordination modes of the fullerene-embedded Cp ring to the (PPh3)Au fragment. For 1 3 (ODCB)2, the cyclopentadienyl ring coordinates to the (Ph3P)Au fragment in an η1 fashion distorted toward η3 geometry, while in 1 3 (CHCl3)2(CS2), crystal packing forces produce enough distor- tion to give near η2 coordination.
Clavaguera, S.; Khan, S. I.; Rubin, Y.* “Unexpected De-Arylation of a Pentaaryl Fullerene”, Org. Lett.2009, 11, 1389-1391.

Abstract: A triphenylamine-derived pentaaryl fullerene undergoes an unusual oxidative dearylation under basic conditions to give tetraarylated epoxy fullerene in high yield. The structure of the product was confirmed by single crystal X-ray diffraction. A mechanism is proposed to account for the loss of the addend and the subsequent formation of the epoxide group.
Kennedy, R. D.; Ayzner, A. L.; Wanger, D. D.; Day, C. T; Halim, M.; Khan, S.I.; Tolbert, S.*; Schwarz, B.*; Rubin, Y.* “Self-Assembling Fullerenes for Improved Bulk-Heterojunction Photovoltaic Devices”, J. Am. Chem. Soc. 2008, 130, 17290–17292.

Abstract: The fullerene adducts 1a and 1b, whose molecular shapes either promote or hinder the formation of 1-D stacks, have been examined for their potential to form 1-D wire-like domains in bulk-heterojunction organic solar cells. The photovoltaic efficiency of solar cells based on blends of the stacking fullerene 1a with regioregular poly(3-hexylthiophene-2,5-diyl) (P3HT) is greatly enhanced compared to nonstacking model fullerene 1b.
Sander, M. Jarrosson, T.; Chuang, S.-C.; Khan, S. I.; Rubin, Y.* “Approaches to Open Fullerenes: Synthesis and Thermal Stability of cis-1 Bis(isobenzofuran) Diels-Alder Adducts of C60”, J. Org. Chem. 2007, 72, 2724 -2731.

Chuang, S.-C.; Sander, M. Jarrosson, T.; James, S.; Rozumov, E. Khan, S. I.; Rubin, Y.* “Approaches to Open Fullerenes: Synthesis and Kinetic Stability of Diels-Alder Adducts of Substituted Isobenzofurans and C60”, J. Org. Chem. 2007, 72, 2716 -2723.

Chuang, S.-C.; Khan, S. I.; Rubin, Y.* “Switch of Electronic Reactivity in Fullerene C60: Activation of Three trans-4 Positions via Temporary Saturation of the cis-1 Positions”, Org. Lett. 2006, 8, 6075-6078.

Chuang, S.-C.; Clemente, F. R.; Khan, S. I.; Houk, K. N.; Rubin, Y.* “Approaches to Open Fullerenes: A 1,2,3,4,5,6-Hexaadduct of C60”, Org. Lett. 2006, 8, 4525-4528.

Clark, A. P.-Z.; Cadby, A. J.; Shen, C. K.-F.; Rubin, Y.*; Tolbert, S. H.* “Synthesis and Self-Assembly of an Amphiphilic Poly(Phenylene Ethynylene) Ionomer”, J. Phys. Chem. B 2006, 110, 22088-22096.
Clark, A. P.-Z.; Shen, K.-F.; Rubin, Y.*; Tolbert, S. H.* “An Amphiphilic Poly(phenylene ethynylene) as the Structure-Directing Agent for Periodic Nanoscale Silica Composite Materials”, Nano Lett., 2005,5, 1647-1652.

Sonmez, G.; Shen, C. K.-F.; Rubin Y.; Wudl, F.* “The Unusual Effect of Bandgap Lowering by C60 on a Conjugated Polymer”, Adv. Mater. 2005, 17, 897-900.

Sonmez, G.; Sonmez, H. B.; Shen, C. K. F.; Jost, R. W.; Rubin, Y.; Wudl, F. “A Processable Green Polymeric Electrochromic”, Macromolecules 2005, 38, 669-675.
Zheng, S.; Thompson, J. D.; Tontcheva, A.; Khan, S. I.; Rubin, Y.* “Perchloro-2,5,8-triazaphenalenyl”,Org. Lett.2005, 7, 1861-1863.

Sonmez, G; Shen, C. K.-F.; Rubin, Y; Wudl, F. “Zero Band Gap and Intrinsic Conductivity in Conjugated Polymers” Polym. Mat. Sci. Engin. 2004, 90, 232-233.
Carravetta, M.; Murata, Y.; Murata, M.; Heinmaa, I.; Stern, R.; Tontcheva, A.; Samoson, A.; Rubin, Y.; Komatsu, K.; Levitt, M. H. “Solid-State NMR Spectroscopy of Molecular Hydrogen Trapped Inside an Open-Cage Fullerene” J. Am. Chem. Soc. 2004, 126, 4092-4093.

Abstract: Solid-state 1H experiments were performed an open-cage fullerene hosting molecular hydrogen. The anisotropy of the molecular hydrogen rotation was studied by double-quantum magic-angle-spinning NMR. The time scale of the molecular hydrogen rotation was estimated by spin-lattice relaxation measurements as a function of temperature.
Sonmez, G., Shen, C. K.-F., Rubin, Y., Wudl, F. “A Red, Green, and Blue (RGB) Polymeric Electrochromic Device (PECD): The Dawning of the PECD Era”, Angew. Chem. Int. Ed. Engl. 2004, 43, 1498-1502.
Shen, C. K.-F.; Sonmez, G.; Smith, A. D.; Schwartz, B. J.; Rubin, Y.; Wudl, F. “Photocurrent Generated with a Homoconjugated Bisfulleroid Polymer” Polymer Preprints 2003, 44, 348-349.
Murata, Y.; Suzuki, M.; Rubin, Y.; Komatsu, K. “Structure of the Hydration Product of the C60-Di(2-pyridyl)-1,2,4,5-tetrazine Adduct”, Bull. Chem. Soc. Jpn., 2003, 76, 1669–1672.
Zheng, S.; Lan, J.; Khan, S. I.; Rubin, Y. “Synthesis, Characterization, and Coordination Chemistry of the 2-Azaphenalenyl Radical”, J. Am. Chem. Soc. 2003, 125, 5786-5791.
Qian, W.; Chuang, S.-C.; Amador, R. B.; Jarrosson, T.; Sander, M.; Pieniazek, S.; Khan, S. I.; Rubin, Y. “Synthesis of Stable Derivatives of C62: The First Nonclassical Fullerene Incorporating a Four-Membered Ring”, J. Am. Chem. Soc.2003, 125, 2066-2067.
Qian, W.; Rubin, Y. “Convergent, Regioselective Synthesis of Tetrakisfulleroids from C60”, J. Am. Chem. Soc.2002, 124, 7683 – 7687.
Asselberghs, I.; Zhao, Y.; Clays, K.; Persoons, A.; Comito, A.; Rubin, Y. “Reversible Switching of Molecular Second-Order Nonlinear Optical Polarizability Through Proton Transfer”, Chem. Phys. Lett.2002, 364, 279-283.
Metzger, R. M.; Baldwin, J. W.; Shumate, W. J.; Peterson, I. R.; Mani, P.; Mankey, G. J.; Morris, T.; Szulczewski, G.; Bosi, S.; Prato, M.; Comito, A.; Rubin, Y. “Electrical Rectification in a Langmuir-Blodgett Monolayer of Dimethylanilinoazafullerene Sandwiched Between Gold Electrodes”, J. Phys. Chem. B2003, 107, 1021-1027.
Irle, S.; Rubin, Y.; Morokuma, K. “ONIOM Study of Ring Opening and Metal Insertion Reactions with Derivatives of C60: Role of Aromaticity in the Opening Process”, J. Phys. Chem. A 2002; 106, 680-688.
Rubin, Y.; Diederich, F. “From Fullerenes to Novel Carbon Allotropes: Exciting Prospects for Organic Synthesis”; in Stimulating Concepts in Chemistry, Vögtle, F.; Stoddart, J. F.; Shibasaki, M.; Eds.; Wiley – VCH, 2001, pp 153-186. (Book chapter)
Rubin, Y.; Jarrosson, T.; Wang, G.-W.; Bartberger, M. D.; Schick, G.; Saunders, M.; Cross, R. J.; Houk, K. N. “Insertion of Helium and Molecular Hydrogen Through the Orifice of an Open Fullerene”, Angew. Chem. Int. Ed. Engl. 2001, 40, 1543-1546.

Abstract: A “molecular surgery” approach has been used to create an opening within a fullerene cage that is large enough to allow atoms and small molecules to pass through. The thermodynamics for the insertion of He and H2 into the open fullerene (left and right pictures, respectively) as well as their escape have been studied by NMR spectroscopy and theoretical methods.

{kind=link}